Cystic Fibrosis: What You Need To Know
- Lilly Scholz

- Apr 9
- 3 min read

What is Cystic Fibrosis?
Cystic Fibrosis is an inherited genetic disorder which causes severe damage mainly to the lungs, digestive system, as well as other vital organs. Normal mucus is slippery and protects the airways and digestive tract. However, cystic fibrosis causes mucus to become thick and sticky, causing blockages, infections, inflammation and even respiratory failure. Cystic Fibrosis is unfortunately a lifelong illness that gets more severe over time. Due to this reason, people with CF often have a shorter life expectancy. However, modern technology and research has led to an improvement to a median age of 44.4 years.
A common misconception is thinking Cystic Fibrosis is a lung disease as it heavily impacts your lungs and airways. However it’s called cystic fibrosis because it also causes cysts and scarring in your pancreas. This damage, along with thick mucus, can block ducts from releasing digestive enzymes, resulting in poor growth, weight loss and malnutrition.
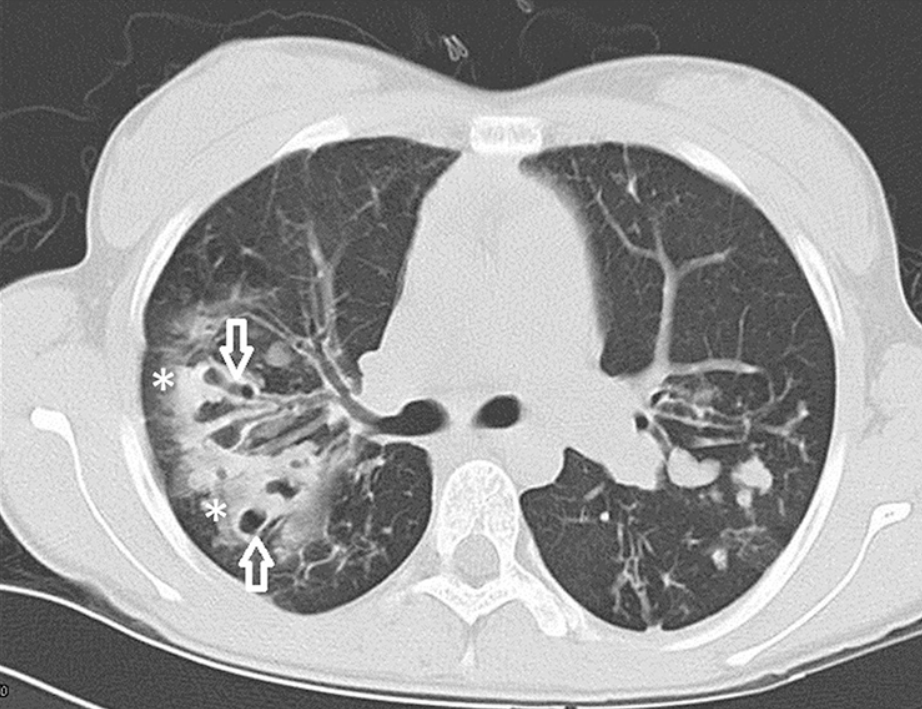
The CT scan above demonstrates the extent to which CF physically damages the lungs. Bronchiectasis, shown by the arrows, refers to permanent widening and damage of the airways, making it hard for the lungs to clear mucus effectively. Moreover, the asterisks indicate mucous plugging. This is when thick, sticky mucus blocks the airways, creating the perfect environment for bacteria to flourish. As a result, patients repeatedly expericne lung infections, inflammation and lung damage.
What Causes Cystic Fibrosis?

Cystic fibrosis is an inherited condition caused by a mutation in the CFTR gene (Cystic Fibrosis Transmembrane Conductance Regulator). In order to have cystic fibrosis, a person must inherit two copies of the mutated CFTR gene, meaning one copy from each biological parent. However, if they only gain one copy this means they will be carriers and could pass the mutated gene to their children, without having cystic fibrosis themselves.
The CFTR gene controls how much salt moves in and out of cells. When the CFTR gene doesn’t work as it should, this results in thick and sticky mucus in mainly the respiratory and digestive tracts, as well as extra salt in sweat.
Symptoms of Cystic Fibrosis:
Respiratory Symptoms:
Chronic cough
Frequent lung infections such as pneumonia or bronchitis
Wheezing and shortness of breath, therefore a reduced ability to exercise
Sinus issues such as stuffy nose
Digestive and Nutritional Symptoms:
Poor growth or difficulty gaining weight
Pancreatitis
Intestinal blockage
Diabetes
Other Potential Symptoms:
Salty sweat
Infertility
Fatigue
Liver disease
Reduced fertility

Treatment and Management of Cystic Fibrosis:
Cystic fibrosis is diagnosed in several ways. For instance, all newborns in the US have to undergo testing for CF, however many people born before 2010 have not been screened. A few drops of blood are taken from the baby’s heel, which is then checked for higher levels of IRT (Immunoreactive trypsinogen) - a chemical released by the pancreas. If the test for IRT comes back positive, the hospital will then likely test the baby’s DNA for CFTR mutations.
Moreover, a sweat test can also be performed to check for high levels of chloride (salt) in your sweat. This test was developed by Lewis Gibson and Robert Cooke in 1959, as chloride levels in sweat are higher for those with CF due to the mutation in the CFTR gene. The table below illustrates how much chloride should be present in a person’s sweat sample to determine whether the diagnosis for CF is positive, unclear or unlikely.

Unfortunately there is no cure for cystic fibrosis, however people with CF are able to manage its symptoms in several ways. The goals of treatment include; preventing and controlling infections that occur in the lungs, removing and loosening mucus from the lungs, getting enough nutrition as well as treating and preventing intestinal blockage.
Respiratory care:
Coughing and breathing techniques: A physical therapist may help teach techniques that open your airways and loosen mucus
Airway clearance vests: This is an inflatable vest that attaches to a machine and vibrates to loosen mucus
Nebulizers: Aerosol mist inhalations via a nebulizer to help open the airways

Nutritional and Digestive care:
Dietary care: A nutritious diet as well as salt and vitamin supplements ensures patients get enough nutrition
Pancreatic Enzyme Capsules: These can be taken with every meal to aid nutrient absorption
Bibliography:
NIH:
Cleveland Clinic:
Cystic Fibrosis Foundation:
Mayo Clinic:


Comments